
離子色譜法(三)
發布時間:2017-09-01
四、電導檢測器
在介紹的HPLC檢測技術同樣適用于離子色譜。在此,僅介紹離子色譜中通用的電導檢測器(CD)。
1、非抑制型電導檢測器
在離子色譜中,分析對象和所使用的流動相都是離子性物質,不同的離子其溶液的導電性是不同的。可以用極限摩爾電導來衡量離子的導電能力(見表13.2)。流動相中主要是淋洗離子和與之平衡的反離子,其電導值稱為背景電導。進樣前,流動相中淋洗離子占領固定相中的離子交換位置。
非抑制型離子色譜使用的是低電導的流動相,如濃度為幾毫摩爾每升的有機酸或有機酸鹽溶液,從色譜柱流出的溶液直接進入電導檢測器。當樣品加入后,樣品帶隨流動相到達色譜柱,被測物質在交換基團上與淋洗離子競爭,達到最初的離子交換平衡,被交換下來的淋洗離子和被測離子的反離子迅速通過色譜柱到達檢測器,在色譜圖上出現一個稱作“水跌”(water dip)的色譜峰(也稱水峰)。各種被測物在色譜柱中的保留不同,依次流出色譜柱,此時流動相中被測離子的濃度增加了,同時有等物質的量(mo1)的淋洗離子交換到了固定相中,由于樣品離子和淋洗離子的摩爾電導不同,這時流動相的電導就不同于背景電導,這種電導的變化就以色譜峰的形式記錄下來。如果淋洗離子的摩爾電導比被測離子小,則在色譜圖上出現正峰,陰離子通常是這種情況。陽離子交換色譜的流動相中的淋洗離子一般是H+,其極限摩爾電導遠比一般陽離子大,所以,陽離子通常產生負峰(可以通過改變電導檢測器的輸出極性得到正峰)。在很多體系中,在被測離子峰之后還會出現一個“系統峰”(system.peak),是由樣品溶液與流動相的組成、pH的差異以及淋洗離子在色譜柱中的保留所引起。它的出現往往給分離或定量帶來負面影響,目前還無法完全消除系統峰,只能設法抑制系統峰的大小和調節系統峰的出峰位置使之對分析無干擾。
2、抑制型電導檢測器
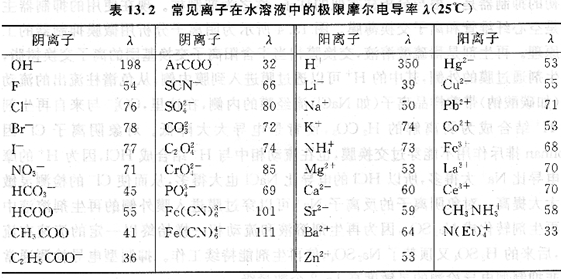
抑制型電導檢測離子色譜使用的是強電解質流動相,如分析陰離子用碳酸鈉、氫氧化鈉,分析陽離子用稀硝酸、稀硫酸等。這類流動相的背景電導高,而且被測離子以鹽的形式存在于溶液中,檢測靈敏度很低,為了提高檢測靈敏度,就需降低背景電導和增加被測離子的電導。分析陰離子時通常用稀硫酸(10~20mmc)l/L)作再生(抑制)劑溶液,分析陽離子時通常用稀氫氧化鈉作再生劑溶液。最初的抑制器是一根與分析柱類似的柱子,稱之為抑制柱。現在使用的抑制器主要是空心纖維管和離子交換薄膜。圖13.4所示為陰離子分析用微膜抑制器的工作原理。再生劑是稀硫酸溶液,交換膜相當于含陽離子交換基團的離子交換樹脂,再生劑通過膜的外側,其中的H+可以透過膜進入到膜內側,從色譜柱流出的流動相(如碳酸鈉)帶著樣品離子(如NaCl)流經膜的內側,在這里,CO2-3與來自再生劑的H+結合成為弱離解的H2CO3,使背景電導大大降低。對象陰離子Cl-因Donnan排斥作用不能穿過交換膜,也在流動相中與H+結合成HCl,因為H+的摩爾電導比Na+大得多,所以HaCl的電導比Na+也大得多,從而使Cl的檢測靈敏度大大提高。對象陰離子的反離子Na。。可以穿過膜進入膜外側的再生劑溶液中使再生劑轉化成Na2S04,因為再生劑溶液和流動相一樣始終以一定的流速在流動,后來的H2S04又頂替了Na2S04,使再生劑能持續工作。抑制型電導檢測通常比非抑制型電導檢測的靈敏度高1~2個數量級。
五、實驗技術
1、去離子水制備及溶液配制
(1)去離子水的制備
用石英蒸餾器制得的蒸餾水的電導率在1μS/cm左右,對于高含量離子的分析,或對分析的要求不高時可以使用。作為一般性要求,離子色譜中使用的純水的電導率應在0.5μs/cm以下。通常用金屬蒸餾器制得的水的電導率在5~25μS/cm,反滲透法(RO)制得的純水電導率在2~40μS/cm,均難以滿足離子色譜的要求。因此需要用專門的去離子水制備裝置制備純水。一般是將以自來水為原水的去離子水再用石英蒸餾器蒸餾,即得到通常所說的重蒸去離子水。也可將RO水作原水引進去離子水制備裝置。精密去離子水制備裝置可以制得電導率O.06μS/cm以下(比電導17MΩ以上)的純水。
2、溶液的配制
配制標準溶液時一定要防止離子污染。樣品溶液和流動相配制好后要用0.5μm以下的濾膜過濾,為防止微生物的繁殖,最好現配現用。
六、流動相的選擇
流動相也稱淋洗液,是用去離子水溶解淋洗劑配制而成。淋洗劑通常都是電解質,在溶液中離解成陰離子和陽離子,對分離起實際作用的離子稱淋洗離子,如用碳酸鈉水溶液作流動相分離無機陰離子時,碳酸鈉是淋洗劑,碳酸根離子才是淋洗離子。選擇流動相的基本原則是淋洗離子能從交換位置置換出被測離子。從理論上講,淋洗離子與樹脂的親和力應接近或稍高于被測離子。但在實際應用中,如果樣品中強保留離子和弱保留離子共存,要是選擇和保留最強的離子的親和力接近的淋洗離子,往往有些弱保留離子很快就流出色譜柱,不能達到分離。因此,合適的流動相應根據樣品的組成,通過實驗進行選擇。
離子抑制色譜除了控制流動相pH值外,對流動相的要求和通常的反相分配色譜一樣。離子對色譜的流動相是由淋洗劑(有機溶劑或水溶液)和離子對試劑組成的。對酸性物質多用季胺鹽(如溴化四甲基銨、溴化四丁基銨、溴化十六烷基三甲基銨)作離子對試劑,對堿性物質則多用烷基磺酸鹽(如己烷磺酸鹽、樟腦磺酸鹽)和烷基硫酸鹽(如十二烷基硫酸鹽)作離子對試劑。離子對試劑的烷基鏈越長,生成的離子對化合物的疏水性越強,在固定相中的保留也隨之增大,但對選擇性的影響不大。所以對于性質很相似的溶質,宜選用烷基鏈較小的離子對試劑。離子對試劑的濃度增大,會促進離子對化合物的形成,所以也會增加被測離子的保留值。但離子對試劑的濃度超過一定的限度后溶質的保留值反而會下降,這可能是由于離子對試劑聚集在樣品離子的周圍增加了配合物在水相中的溶解度的緣故。所以離子對試劑有一個合適的濃度范圍,較小的離子對試劑應在10mmol/L以下,較大的離子對試劑(碳鏈在12以上)在5mmol/L以下。
反相離子對色譜中用得最多的流動相也是甲醇一水體系,因為甲醇對許多離子對試劑有較好的溶解性。乙腈-水體系柱效也很高,但對離子對試劑的溶解性不是太好。
對分離影響較大的另一個因素是流動相的pH值,它決定被測物質的解離程度。對于硅膠基質的鍵合固定相,流動相的pH值應為2~8。某些緩沖劑離子也有可能與離子對試劑結合,所以緩沖劑的濃度不宜過高,通常為1~5mmol/L。
1、定性方法
當色譜柱、流動相及其他色譜條件確定后,便可根據分離機理和經驗知道哪些離子在這個條件下有可能保留,而且還能根據離子的性質大致判斷其保留順序。在此基礎上,就可以用標準物質進行對照。在確定的色譜條件下保留時間也是確定的,與標準物質保留時間一致就可初步認為是這種離子。這種方法稱作保留時間定性法。
很多離子具有選擇性或專屬性顯色反應,也可以用顯色反應進行定性。
質譜的定性能力很強,如果離子色譜和質譜聯用就可以很準確地定性。與I.C―MS聯用技術一樣,LC―MS聯用也是在接口上存在一些困難,加上儀器昂貴,在我國尚未得到應用。
2、定量方法
在一定的被測物濃度范圍內,色譜峰的高度和面積與被測離子濃度成線性關系,但一般情況下面積工作曲線的線性范圍要寬一些,所以通常以峰面積的大小進行定量。IC定量方法與其他分析方法一樣。用得最多的是標準曲線法(1點或多點)、標準加入法和內標法。
參考資料:現代儀器分析實驗與技術






