
電分析化學法儀器結構與原理(二)
發(fā)布時間:2017-09-01
三、實驗技術
1、電位分析實驗技術
直接電位分析法可用標準曲線法和標準加入法。標準曲線法是在同樣的條件下由標準物質配制一系列不同濃度的標準溶液,由其濃度的對數(shù)與電位值作圖求得校準曲線,再在相同條件下測定試樣溶液的電位值,由校準曲線上讀取試樣中待測離子含量。校準曲線法的缺點是當試樣組成復雜時,難以保證其組成與校準曲線的條件完全一致,因而有時需要由加人回收實驗對方法的準確性加以驗證。標準加入法是將一定體積和濃度的標準溶液加入到已知體積的試樣中,根據(jù)加入前后電位變化計算試液中待測離子濃度。標準加入法的優(yōu)點是標準溶液和待測溶液中的被測離子是在非常接近的條件下測定的,因而測定結果更加可靠。利用惰性金屬如鉑電極作指示電極,用飽和甘汞電極作參考電極,可指示體系滴定過程中的氧化還原電極電位的變化。在化學計量點附近產(chǎn)生電位突躍而指示到達終點。
2、電解分析實驗技術
在庫侖滴定中電解電流是恒定的,因此只要準確測定滴定開始至終點所需要的時間,就可準確測定被滴定物的量。準確地指示滴定終點是非常重要的,指示終點的方法有化學指示劑法、電位法、雙鉑電極法等。雙鉑電極法又稱永停法,其在碘庫侖滴定法中指示終點的原理為:在兩鉑片電極之間加10~200mV的小電壓,在滴定終點之前,電解產(chǎn)生的I2全部與AsO3-3反應,溶液中僅有極少量I2存在,而AsO3-3和AsO3-4大量存在。因而溶液電極電位主要由電對AsO3-3/AsO3-4確定,但AsO3+4+2e=AsO3-3電對為不可逆電對,兩個電極間加小電壓不能產(chǎn)生電流。相反,電對I2+2e=2I一為可逆電對,當?shù)味ǖ竭_終點時,一旦溶液中有略過量的I2出現(xiàn),立即在電路中產(chǎn)生一電解電流。因此一旦指示電路中出現(xiàn)電流,表明終點到達。
四、極譜和伏安分析實驗技術
1、懸汞電極和汞膜電極制備
懸汞電極使用方便,在商品的滴汞電極的汞流路上裝有電磁閥,可自動產(chǎn)生大、中、小3種不同體積的懸汞滴。使用擠壓式懸汞電極時,旋轉千分尺推動頂針擠壓儲汞器中的汞,使汞從與儲汞器相連的毛細管流出形成汞滴,汞滴的大小由千分尺旋轉的刻度控制。掛汞式懸汞電極是將直徑為0.1mm的鉑絲、金絲或銀絲封在一適當直徑的玻璃管的一端,露出部分的長度約O.1mm,另一端用汞接觸的方法引出導線。將此電極浸人飽和硝酸亞汞的硝酸溶液中(0.2~0.5mol/L),在25~30mA的電流下電解1~3min,使汞沉積在鉑絲上,可制得直徑為1.O~1.5mm的懸汞滴。或將上述制得的電極外端磨平,除去表面的氧化膜,電解鍍上一層汞,然后掛上一定大小的汞滴,即成為掛汞式懸汞電極。
在固體電極如玻碳、銀或鉑電極表面鍍上一層薄的汞膜,即為汞膜電極。使用較為普遍的為玻碳汞膜電極。在絕緣管的一端封接一玻碳薄片,另一端接出導線。先將其表面在稀的汞鹽溶液中電解鍍上一層汞膜,然后插入試液中使用,或者在試液中加入少量汞鹽,在電解富集過程中與被測物同時在玻碳上析出形成汞膜和汞齊(同位鍍汞)。汞膜的厚度可由溶液中汞鹽濃度和電解時間來控制。
2、固體電極表面處理
固體電極處理的第一步是進行機械研磨、拋光至鏡面程度。通常用于拋光電極的材料有金剛砂、CeO2、ZrO、MgO和a-A12 O3粉及拋光液。拋光時按拋光劑粒度降低的順序依次進行研磨,對新的電極表面應先經(jīng)金剛砂紙粗磨和細磨后,再用a-Al2O3粉按照1.O,0.3,O.05μm粒度在平板玻璃或拋光布上分別進行拋光。每次拋光后先洗去表面污物,再移入超聲水浴中清洗,每次2~3min,重復3次。最后用乙醇、稀酸和水徹底洗滌,得到一個平滑光潔、新鮮的電極表面。
固體電極經(jīng)拋光后接著進行化學的或電化學的處理,尤其電化學處理,是最常用的清潔、活化電極表面的手段。電化學處理常用強酸或中性電解質溶液,有時也用具有弱的絡合性的緩沖溶液在恒電位、恒電流或循環(huán)電位掃描下極化,根據(jù)掃描電位終止的電位不同,可獲得氧化的、還原的或干凈的電極表面。電化學處理方法還能在試液中直接進行電極處理,方法簡單易行。
3、除氧
電解液中溶解的微量O2在室溫下可達8mg/L。在極譜分析時,O2也在汞電極上還原,產(chǎn)生兩個極譜波:
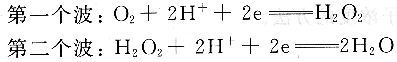
第一個氧波的半波電位約為一0.2V,第二個氧波的半波電位約為一O.8V,均為不可逆的寬帶波,干擾測定。因此電化學實驗前試液必須除氧,方法是向溶液中通高純氮l~2min。精確測量時為了不影響試液的濃度,氮氣要用溶劑蒸氣進行預飽和。測試過程中停止通氮氣,但試液要保持在氮氣氛圍中。
在中性或堿性溶液中也可通過加亞硫酸鈉除氧,使氧與之反應生成sO2-4:
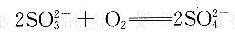
4、超痕量分析的試劑純化
在極譜和伏安分析時降低或消除雜質的影響是獲得高準確度結果的重要條件。要消除殘余電流需選用高純度水及試劑作底液,如仍不滿足,通過控制電位汞陰極電解法可除去底液中存在的可還原雜質,濃度可低至O.1μg/L以下。汞的純度也影響殘余電流,特別是陽極溶出分析。汞可用洗滌法或蒸餾法提純。但汞是易揮發(fā)的有毒物質,蒸餾汞時應在密封容器中進行,并在通風良好的場所進行。
5、極譜極大的消除
極譜極大是極譜分析中常見的一種現(xiàn)象。電解開始時,電流隨電壓的增加迅速增加,達到一個極大值,然后再下降至擴散電流的數(shù)值。這種在擴散電流之前出現(xiàn)的峰值電流比擴散電流大許多,干擾極譜的正常分析,稱為“極譜極大”現(xiàn)象。這是因為在極譜過程中,汞滴上部由于被毛細管末端所遮蔽,可還原離子不易接近汞表面,因而電流密度較小;而汞滴下部可還原離子可以無阻礙接近汞滴,因而電流密度較大。電流密度不均勻,導致滴汞表面附近的表面張力不一樣,從而導致汞滴表面溶液的切向運動,可還原離子便因攪動而更快地達到電極表面,使極譜波產(chǎn)生極大現(xiàn)象。向溶液中加入微量的表面活性物質,例如,用量不超過溶液的O.1%的動物膠、Triton X-100、甲基紅等,便可消除極譜極大現(xiàn)象。
五、實驗
1、氟離子選擇性電極測定水中氟離子含量
(1)實驗目的
學習用直接電位法測定氟離子濃度的方法。
(2)實驗原理
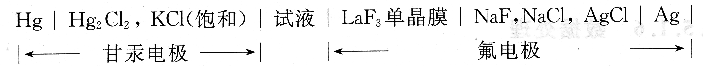
在離子強度和pH值不變時,整個電池的電動勢為:
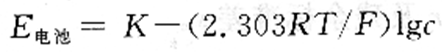
由上式可見,電池電動勢與試液中的氟離子濃度的對數(shù)呈線性關系。
本實驗采用標準曲線法進行定量。
(3)儀器與試劑
儀器:帶微處理器的離子計、氟離子選擇電極、單液接參考電極、電磁攪拌器。
試劑:NaF標準溶液:0.1000mol/L;
總離子強度調節(jié)緩沖溶液(TISAB):溶解58.8g檸檬酸鈉和20.2g KNO3于少量水中,加800mL水,以HCl或NaOH調節(jié)pH值至6.5,稀釋至1L;1.0×10-31mol/L NaF溶液。
2、實驗內(nèi)容與步驟
(1)氟電極準備。氟電極在使用前于1.0×10-3mol/L NaF溶液中浸泡活化1~2h。用去離子水清洗電極,并測量其電位至與去離子水中的電位值相接近(約一300mV)。
(2)預熱儀器約20min,接入氟電極與參考電極。
(3)校準曲線繪制。由0.1000mol/L標準NaF溶液配制一系列NaF標準溶液各50mL。其中各含25mL總離子強度調節(jié)緩沖液和10-2,10-3,10-4,10-5,10-6 m01/L F一。將上述溶液倒入洗凈并干燥的50mL燒杯中,放入磁攪拌子,插入電極。在離子計上按由稀至濃的順序測定不同F(xiàn)一濃度的電位值,記下讀數(shù)。測定時攪拌2min,靜置lmin,待電位穩(wěn)定后讀數(shù)。以測得的毫伏數(shù)為縱坐標,以F-濃度的對數(shù)為橫坐標作校準曲線。
(4)水中氟離子濃度的測定。往燒杯中準確移取25.00mL水樣,加25.00mL總離子強度調節(jié)緩沖液。用離子計測定電位值,重復3次。
(5)清洗電極。實驗結束后,用去離子水清洗電極至電位值與起始空白電位值相近,收入電極盒中保存。
3、注意事項
電極電位在攪拌時和靜止時讀數(shù)不同,測定過程中讀數(shù)狀態(tài)應保持一致。
4、數(shù)據(jù)處理
(1)在校準曲線的線性區(qū)間,用最小二乘法進行曲線擬合,計算校準曲線的斜率k、截距b、相關系數(shù)r及殘余標準差s。
(2)計算未知樣中F-濃度的平均值(mol/L)及標準偏差。
參考資料:現(xiàn)代儀器分析實驗與技術






